As I've mentioned in the past, my PhD project is involved with using high-throughput sequencing (HTS) to investigate T-cell receptor (TCR) repertoires.
Next-generation sequencing (NGS) technologies allow us to investigate the workings and dynamics of the adaptive immune system with greater breadth and resolution than ever before. It’s possible to extract or amplify the coding sequence for thousands to millions of different clones, and throw them all into one run of a sequencer. The trick is extracting the TCR information from the torrent of data that pours out.
Our lab’s paper for the high-throughput analysis of such repertoire data has been out for a while now, so here’s my attempt to put in to a little wider context than a paper allows, while hopefully translating it for the less computationally inclined.
In order to understand best how the brains in our lab tackled this problem, it's probably worth looking at how others before us have.
There's a beaten path to follow. The germline V, D and J gene sequences are easily downloaded from
IMGT GENE-DB; then you take your pick of a short read aligner to match up the V and J sequences used (we’ll come to the Ds later).
Most of the popular alignment algorithms get covered by some group or other:
SWA (Ndifon et al., 2012; Wang et al., 2010),
BLAT (Klarenbeek et al., 2010) and
BLAST (Mamedov et al., 2011) all feature. IMGT’s
HighV-QUEST software uses DNAPLOT, a bespoke aligner written for their previous low-throughput version (Alamyar et al, 2012; Giudicelli et al, 2004; Lefranc et al, 1999).
Sadly, some of the big hitters in the field don’t see fit to mention what they use to look for TCR genes (or I’ve just missed it).
Robert Holt’s group produced the first NGS TCR sequencing paper I’m aware of (Freeman, Warren, Webb, Nelson, & Holt, 2009), but don’t mention how they assign their genes (admittedly they’re more focused on explaining how
iSSAKE, their short-read assembler designed for TCR data, works).
The most prolific author in the TCR ‘RepSeq’ field is
Harlen Robins, who has contributed to a wealth of wonderful repertoire papers (Emerson et al., 2013; Robins et al., 2009, 2010, 2012; Sherwood et al., 2013; Srivastava & Robins, 2012; Wu et al., 2012), yet all remain equally vague on TCR assignation methods (probably related to the fact that he and several other early colleagues set up a company,
Adaptive Biotech, that offers TCR repertoire sequencing and analysis).
So we see a strong tendency towards alignment (and a disappointing prevalence of ‘in house scripts’). I can understand why: you've sequenced your TCRs, you've got folder of fastqs burning a hole in your hard drive, you’re itching to get analysing. What else does your average common house or garden biologist do when confronted with sequences, but align?
However, this doesn't really exploit the nature of the problem.
When trying to fill out a genome, alignment and assembly make sense; you take a read, and you either see where it matches to a reference, or see where it overlaps with the other reads to make a contig.
For TCR analysis however, we're not trying to map reads to or make a genome; arguably we’re dealing with some of the few sequences that might not be covered under the regular remit of 'the genome'. Nor are we trying to identify known amplicons from a pool, where they should all the same, give or take the odd SNP.
TCR amplicons instead should have one of a limited number of sequences at each end (corresponding to the V and J/C gene present, depending on your TCR amplification/capture strategy), with a potentially (and indeed probably) completely novel sequence in between.
In this scenario, pairwise alignment isn’t necessarily the best option. Why waste time trying to match every bit of each read to a reference (in this case, a list of germ-line V(D)J gene segments), when only the bits you’re least interested in – the germline sequences – stand a hope of aligning anywhere?
Enter our approach:
Decombinator (Thomas, Heather, Ndifon, Shawe-Taylor, & Chain, 2013).
Decombinator rapidly scans through sequence files looking for rearranged TCRs, classifying any it finds by these criteria; which V and J genes were used, how many nucleotides have been deleted from each, and the string of nucleotides between the end of the V and the start of the J. This simple five-part index condenses all of the information contained in any given detected rearrangement into a compact format, convenient for downstream processing.
All TCR analysis probably does something similar; the clever thing about Decombinator is how it gets there. At its core lies a
finite state automaton that passes through each read looking for a panel short sequence ‘tags’, each one uniquely identifying the presence of a germline V or J gene. It if finds tags for both a V and a J it can populate the five fields of the identifier, thus calling the re-arrangement.
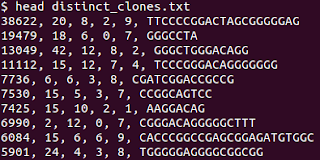 |
| Example of the comma-delimited output from Decombinator. From left to right: (optional sixth-field) clonal frequency; V gene used; J gene used; number of V deletions; number of J deletions, and insert string |
The algorithm used was
developed by Aho and Corasick in the ‘70s, for bibliographic text searching, and the computer science people tell me that it’s really never been topped – when it comes to searching for multiple strings at once,
the Aho-Corasick (AC) algorithm is the one (Aho & Corasick, 1975).
Its strength lies in its speed – it’s simply the most effective way to search one target string for multiple substrings. By using the AC algorithm Decombinator runs orders of magnitude faster than alignment based techniques. It does this by generating a special
trie of the substrings, which it uses to search the target string exceedingly efficiently.
Essentially, the algorithm uses the trie to look for every substring or tag simultaneously, in just one pass through the sequence to be searched. It passes through the string, using the character at the current position to decide how to navigate; by knowing where it is on the trie at any one given point, it’s able to use the longest matched suffix to find the longest available matched prefix.
Figshare seems to have scotched up the resolution somewhat, but you get the idea.
Decombinator uses a slight modification of the AC approach, in order to cope with mismatches between the tag sequences and the germline gene sequenced (perhaps from SNPs, PCR/sequencing error, or use of non-prototypic alleles). If no complete tags are found, the code breaks each of the tags into two halves, making new half-tags which are then used to make new tries and search the sequence.
If a half-tag is found, Decombinator then compares the sequence in the read to all of the whole-tags that contain that half-tag
*; if there’s only a one nucleotide mismatch (a
Hamming distance of one) then that germline region is assigned to the read. In simulated data, we find this to work pretty well, correctly assigning ~99% of artificial reads with a 0.1% error (i.e. one random mismatch every 1,000 nucleotides, on average), dropping to ~88% for 1% error
**.
It’s simple, fast, effective and open source; if you do have high-throughput human or mouse alpha-beta or gamma-delta TCR data set to analyse, it’s probably worth giving it a try. The only real available other option is HighV-QUEST, which (due to submission and speed constraints) might not be too appealing an option if you really have a serious amount of data.
(
CORRECTION – in the course of writing this entry (which typically has gone on about five times longer than I intended, in both time taken and words written), some new rather exciting looking software has just been published (Bolotin et al., 2013).
MiTCR makes a number of bold claims which if true, would make it a very tempting bit of software. However, given that I haven’t been able to get it working, I think I’ll save any discussion of this for a later blog entry.)
D segments
If you’ve made it this far through the post, chances are good you’re either thinking about doing some TCR sequencing or have done already, in which case you’re probably wondering, ‘but what about the Ds – when do they get assigned’?
In the beta locus (of both mice and men), there are but two D regions, which are very short, and very similar. As they can get exonucleolytically nibbled from both ends, the vast majority of the time you simply can’t tell which has been used (one early paper managed in around a quarter of reads (Freeman et al., 2009)). Maybe you could do some kind of probabilistic inference for the rest, based on the fact that there is a correlation between which Ds pair with which Js, likely due to the chromatin conformation at this relative small part of the locus (Murugan, Mora, Walczak, & Callan, 2012; Ndifon et al., 2012), but that’s a lot of work for very little reward.
Hence Decombinator does not assign TRBDs; they just get included in the fifth, string based component of the five-part identifier (which explains the longer inserts you see for beta compared to alpha). If you want to go TRBD mining you’re very welcome, just take the relevant column of the output and get aligning. However, for our purposes (and I suspect many others’), knowing the exact TRBD isn’t that important, where indeed even possible at all.
Errors
There’s also the question of how to deal with errors, which can accrue during amplification or sequencing of samples. While Decombinator does mitigate error somewhat through use of the half-tags and omission of reads containing ambiguous N calls, it doesn’t have any other specific error-filtration. As with any pipeline, garbage in begets garbage out; there’s plenty of software to trim or filter HTS data, so we don’t really need to reinvent the wheel and put some in here.
Similarly, Decombinator doesn’t currently offer sequence clustering, whereby very similar sequences get amalgamated into one, as some published pipelines do. Personally, I have reservations about applying standard clustering techniques to variable immunoreceptor sequence data.
Sequences can legitimately differ by only one nucleotide, and production of very similar clonotypes is inbuilt into the recombination machinery (Greenaway et al., 2013; Venturi et al., 2011); it is very easy to imagine bona fide but low frequency clones being absorbed into their more prevalent cousins, which could obscure some genuine biology. The counter argument is of course that by not clustering, one allows through a greater proportion of errors, thus artificially inflating diversity. Again, if desired
other tools for sequence clustering exist.
Disclaimer
My contribution to Decombinator was relatively minor - the real brainwork was done before I'd even joined the lab, by my labmate, mathematician
Niclas Thomas, our shared immunologist supervisor
Benny Chain, and Nic's mathematical supervisor,
John Shawe-Taylor. They came up with the idea and implemented the first few versions. I came in later, designing tags for the human alpha chain, testing and debugging, and bringing another biologist’s view to the table for feature development
***. The other author,
Wilfred Ndifon, at the time was a postdoc in a group with close collaborative ties, who I believe gave additional advice on development, and provided pair-wise alignment scripts against which to test Decombinator.
* Due to the short length of the half-tags and the high levels of homology between germline regions, not all half tags are unique
** In our Illumina data, by comparing the V sequence upstream of the rearrangement – which should be invariant – to the reference, we typically get error rates below 0.5%, some of which could be explained by allelic variation or SNPs relative to the reference
*** At first we had a system that Nic would buy a drink for every bug found. For a long time I was the only person really using Decombinator (and probably remain the person who’s used it most), often tweaking it for whatever I happened to need to make it do that day, putting me in prime place to be Bug-Finder Extraordinaire. I think I let him off from the offer after the first dozen or so bugs found.
Aho, A. V., & Corasick, M. J. (1975). Efficient string matching: an aid to bibliographic search. Communications of the ACM, 18(6), 333–340. doi:10.1145/360825.360855
Alamyar, A., Giudicelli, V., Li, S., Duroux, P., & Lefranc, M. (2012). IMGT/HIGHV-QUEST: THE IMGT® WEB PORTAL FOR IMMUNOGLOBULIN (IG) OR ANTIBODY AND T CELL RECEPTOR (TR) ANALYSIS FROM NGS HIGH THROUGHPUT AND DEEP SEQUENCING. Immunome Research, 8(1), 2. doi:10.1038/nmat3328
Bolotin, D. a, Shugay, M., Mamedov, I. Z., Putintseva, E. V, Turchaninova, M. a, Zvyagin, I. V, Britanova, O. V, et al. (2013). MiTCR: software for T-cell receptor sequencing data analysis. Nature methods, (8). doi:10.1038/nmeth.2555
Emerson, R., Sherwood, A., Desmarais, C., Malhotra, S., Phippard, D., & Robins, H. (2013). Estimating the ratio of CD4+ to CD8+ T cells using high-throughput sequence data. Journal of immunological methods. doi:10.1016/j.jim.2013.02.002
Freeman, J. D., Warren, R. L., Webb, J. R., Nelson, B. H., & Holt, R. a. (2009). Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome research, 19(10), 1817–24. doi:10.1101/gr.092924.109
Giudicelli, V., Chaume, D., & Lefranc, M.-P. (2004). IMGT/V-QUEST, an integrated software program for immunoglobulin and T cell receptor V-J and V-D-J rearrangement analysis. Nucleic acids research, 32(Web Server issue), W435–40. doi:10.1093/nar/gkh412
Greenaway, H. Y., Ng, B., Price, D. a, Douek, D. C., Davenport, M. P., & Venturi, V. (2013). NKT and MAIT invariant TCRα sequences can be produced efficiently by VJ gene recombination. Immunobiology, 218(2), 213–24. doi:10.1016/j.imbio.2012.04.003
Klarenbeek, P. L., Tak, P. P., Van Schaik, B. D. C., Zwinderman, A. H., Jakobs, M. E., Zhang, Z., Van Kampen, A. H. C., et al. (2010). Human T-cell memory consists mainly of unexpanded clones. Immunology letters, 133(1), 42–8. doi:10.1016/j.imlet.2010.06.011
Lefranc, M.-P. (1999). IMGT, the international ImMunoGeneTics database. Nucleic acids research, 27(1), 209–212. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=165532&tool=pmcentrez&rendertype=abstract
Mamedov, I. Z., Britanova, O. V, Bolotin, D., Chkalina, A. V, Staroverov, D. B., Zvyagin, I. V, Kotlobay, A. A., et al. (2011). Quantitative tracking of T cell clones after hematopoietic stem cell transplantation. EMBO molecular medicine, 1–8.
Murugan, a., Mora, T., Walczak, a. M., & Callan, C. G. (2012). Statistical inference of the generation probability of T-cell receptors from sequence repertoires. Proceedings of the National Academy of Sciences. doi:10.1073/pnas.1212755109
Ndifon, W., Gal, H., Shifrut, E., Aharoni, R., Yissachar, N., Waysbort, N., Reich-Zeliger, S., et al. (2012). Chromatin conformation governs T-cell receptor J gene segment usage. Proceedings of the National Academy of Sciences, 1–6. doi:10.1073/pnas.1203916109
Robins, H. S., Campregher, P. V, Srivastava, S. K., Wacher, A., Turtle, C. J., Kahsai, O., Riddell, S. R., et al. (2009). Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood, 114(19), 4099–107. doi:10.1182/blood-2009-04-217604
Robins, H. S., Desmarais, C., Matthis, J., Livingston, R., Andriesen, J., Reijonen, H., Carlson, C., et al. (2012). Ultra-sensitive detection of rare T cell clones. Journal of Immunological Methods, 375(1-2), 14–19. doi:10.1016/j.jim.2011.09.001
Robins, H. S., Srivastava, S. K., Campregher, P. V, Turtle, C. J., Andriesen, J., Riddell, S. R., Carlson, C. S., et al. (2010). Overlap and effective size of the human CD8+ T cell receptor repertoire. Science translational medicine, 2(47), 47ra64. doi:10.1126/scitranslmed.3001442
Sherwood, A. M., Emerson, R. O., Scherer, D., Habermann, N., Buck, K., Staffa, J., Desmarais, C., et al. (2013). Tumor-infiltrating lymphocytes in colorectal tumors display a diversity of T cell receptor sequences that differ from the T cells in adjacent mucosal tissue. Cancer immunology, immunotherapy: CII. doi:10.1007/s00262-013-1446-2
Srivastava, S. K., & Robins, H. S. (2012). Palindromic nucleotide analysis in human T cell receptor rearrangements. PloS one, 7(12), e52250. doi:10.1371/journal.pone.0052250
Thomas, N., Heather, J., Ndifon, W., Shawe-Taylor, J., & Chain, B. (2013). Decombinator: a tool for fast, efficient gene assignment in T-cell receptor sequences using a finite state machine. Bioinformatics, 29(5), 542–50. doi:10.1093/bioinformatics/btt004
Venturi, V., Quigley, M. F., Greenaway, H. Y., Ng, P. C., Ende, Z. S., McIntosh, T., Asher, T. E., et al. (2011). A Mechanism for TCR Sharing between T Cell Subsets and Individuals Revealed by Pyrosequencing. Journal of immunology. doi:10.4049/jimmunol.1003898
Wang, C., Sanders, C. M., Yang, Q., Schroeder, H. W., Wang, E., Babrzadeh, F., Gharizadeh, B., et al. (2010). High throughput sequencing reveals a complex pattern of dynamic interrelationships among human T cell subsets. Proceedings of the National Academy of Sciences of the United States of America, 107(4), 1518–23. doi:10.1073/pnas.0913939107
Wu, D., Sherwood, A., Fromm, J. R., Winter, S. S., Dunsmore, K. P., Loh, M. L., Greisman, H. a., et al. (2012). High-Throughput Sequencing Detects Minimal Residual Disease in Acute T Lymphoblastic Leukemia. Science Translational Medicine, 4(134), 134ra63–134ra63. doi:10.1126/scitranslmed.3003656