I covered an interesting (if slightly intricate) paper for journal club last week, so I thought I'd go over some of the major techniques and findings here. Note that while I'm going to try to keep it simple, (for the purposes of this post) I'm going to assume a rudimentary knowledge of immunology.
First off,
here is the paper, that came out in
last month's edition of Immunity, entitled "Subsets of Nonclonal Neighboring CD4+ T Cells Specifically Regulate the Frequency of Individual Antigen-Reactive T Cells"
1.
Coming from the group of
Ronald Schwartz at the NIH, this body of work is interested in the mechanisms of T cell homeostatis.
Let's get fundamental. Our bodies are of a finite size, so we can only fit so many cells in our circulatory systems and tissues. In terms of our T cells, we want to keep as wide a repertoire of different T cell receptor (TCR) bearing clones as possible, to maximise the number of potential pathogens and challenges we can recognise.
However, after a T cell gets activated by its cognate pMHC target (and has expanded and differentiated into effector cells to clear the challenge), how does our body get rid of all those expanded clones, contracting that population and
only that population?
This work builds on
previous work from the same lead author, Nevil Singh, where they noticed something interesting
2.
In order to understand what's gone on, I just need to explain a few of the basics of their model systems.
Both papers make heavy use of adoptively transferring transgenic-TCR bearing T cells, into mice which either have their own endogenous T cell machinery in place, or who are T cell deficient (CD3
ε knock outs).
The main cell they transfer are 5C.C7 T cells, which bear a TCR that recognises a peptide of PCC (pidgeon cytochrome C). This group's previous paper found that when transferred into mice that express PCC (under a MHC-I promoter), one of two things can happen.
If the recipient mouse lacks its own, functioning T cell system (PCC
+/Cd3e-/-) then the transferred 5C.C7 T cells persist in that mouse, causing an autoimmune arthritis.
If the recipient mouse has it's own T cells however (PCC
+/
Cd3e+/+), then the 5C.C7 T cells not only fail to cause any disease, but they actually disappear from that mouse over time. Or, as they would have it, the 5C.C7 cells get 'deleted'.
At the start of this recent paper, they recapitulate this finding in a neat experiment where they transfer
5C.C7 cells (from transgenic, Rag2-/- mice3) into PCC
+/Cd3e-/- mice, under one of three conditions; either alone, or having received a million CD4+ or CD8+ polyclonal T cells the day before (figure 1A).
It seems that the deletor activity they'd noticed previously is entirely due to some component of a polyclonal CD4+ response.
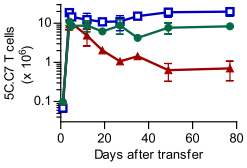 |
| Figure 1A (from the paper1): Survival of 5C.C7 T cells transferred into a T cell deficient mouse (blue line), compared against mice who received one million CD8+ (green line) or CD4+ (red line), measured in pooled lymph node and spleen. 5C.C7 cells measured by double staining for CD4 and Vβ3, the beta variable gene used in the 5C.C7 TCR. |
However, when they go on to try and narrow down what CD4+ T cell might be responsible, the deletor phenotype doesn't appear to easily fractionate to any obvious subset.
Without a clear smoking gun, they adopt an alternate tack. They knew that transferring a million CD4 T cells into the target mouse protected them from the persistence and pathology of the 5C.C7 cells. So, they thought to try and divide this million up, and see which fractions (if any) remain deletors.
To do this they flow sort polyclonal CD4+ T cells into random pools of 100 cells, which they call 'centipools'. Each centipool is then expanded in vitro, and then a million of each expanded population is transferred into a mouse to assay for 5C.C7 deletor activity.
They found that of the 54 centipools tested, there were three that seemed to recapitulate the entire deletor phenotype.
They probed these pools all manor of ways to see if they could pin down what was responsible. They ran microarrays and sequenced TCR genes; nothing seemed to correlate with deletor ability, and nor was any one TCR clone shared across all three centipools.
With no hook to hang a hypothesis on, they go on the attack, and simply take all the TCR sequences from the strongest-deletor centipool and generate
retrogenic TCR T cells for each of them.
Transferring these retrogenic T cells into the PCC+, T cell deficient mice reveals that within a given centipool, there's only one clone that's reponsible for
all of the deletor phenotype. Moreover, this clone deletes 5C.C7 even in PCC- mice; there doesn't need to be target antigen present!
The authors then turn back to the literature; 5C.C7 is a well-studied TCR, perhaps there's something in the back catalogue that might be pertinent. Lo and behold, it turns out there are other peptides to which 5C.C7 is known to react, including one from the endogenous retrovirus GP, which is implicated in
positively selecting 5C.C7 in the thymus
4.
There follows some fairly fiddly experiments (which probably contain the least convincing data in the paper), where they reportedly show that this GP-derived peptide is actually a shared ligand for both the 5C.C7 TCR and the retrogenic TCR cloned from the deletor centipool.
You have to pump the mice full of GP peptide, but it seems that eventually you do see a slight proliferation of both 5C.C7 and deletor T cell, a feature which interestingly could not be repeated in vitro. Apparently, all other assays failed to assign the GP peptide as a stimulating ligand for either cell type.
So, show me the model.
The idea is that different, unrelated T cell clones can be categorised into colonies, based on shared, sub-threshold ligands (e.g. the 5C.C7 and deletor cells described in the paper).
If a member of a colony gets expanded by its (above threshold) ligand, it will expand. However, the contraction phase will then be mediated at the colony-level, enforced by other clonotypes within that colony, preventing collateral damage to non-colonial T cells and thus maintaining a rich, diverse TCR repertoire.
I really quite like this paper; it was a fiddly finding to unravel, and they make use of a number of techniques to do so. I'm not 100% convinced about the GP peptide, but should it be true it's an incredibly intriguing finding. It also would have been nice if they'd made inroads into finding out the mechanism of this colony-regulation, but I suppose there's always the next paper.
My PhD project is all about sequencing TCR repertoires, and day-to-day we tend to operate one the naive assumption that by-and-large the information required to maintain a given T cell clone's population is wrapped up in the genotype and phenotype of that clone. The idea that every T cell clonotype could be modulated by several others within its colony (and in turn be modulating that colony's other members) is fascinating, and could well represent another level of intricate regulation to a marvellously complex biological system.
For an alternative write up of this paper, this edition of the journal
also featured a preview of the piece.
If you're interested, you can also check out the
prezi presentation I gave in my journal club presentation of this paper.
1. Singh, N. J., Bando, J. K., & Schwartz, R. H. (2012). Subsets of Nonclonal Neighboring CD4(+) T Cells Specifically Regulate the Frequency of Individual Antigen-Reactive T Cells. Immunity, 37(4), 735-46. Elsevier Inc. doi:10.1016/j.immuni.2012.08.008 ($)
2. Singh, N. J., Chen, C., & Schwartz, R. H. (2006). The impact of T cell intrinsic antigen adaptation on peripheral immune tolerance. PLoS biology, 4(11), e340. doi:10.1371/journal.pbio.0040340
3. As these are Rag2 knock outs they would be unable to undergo endogenous V(D)J rearrangement, and thus will only produce mature T cells for the transgenic 5C.C7 receptor
4. Ebert, P.J., Jiang, S., Xie, J., Li, Q.J., and Davis, M.M. (2009). An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nat. Immunol. 10, 1162–1169. doi:10.1038/ni.1797 ($)




